Drug discovery has always been a balancing act, with scientists constantly striving for ‘Goldilocks’ compounds – the search for molecules that achieve on-target activity, while also avoiding hidden liabilities such as toxicity, poor exposure, or unfavorable pharmacokinetics, can seem insurmountable at times. These failures, often discovered downstream, can cost discovery programs years of effort and millions of dollars.
Part of the challenge stems from how discovery programs have been historically segmented. Molecular design efforts in one team, ADMET assessment and modeling in another, with pharmacokinetic (PK) considerations often deferred until much later, often after synthetic resources have already been invested. Such separation can slow iteration and obscure early warning signs.
With the continued advancement of in silico modeling, the opportunity to unify these stages is now. By integrating ADMET and PK awareness into the earliest design iterations, discovery chemists can identify and prioritize compounds that are both active and viable, streamlining the Design-Make-Test-Analyze (DMTA) cycle and reducing late-stage attrition.
Through ADMET Predictor®, this integrated approach within a singular platform can be summarized as a one that supports a Molecular Discovery – ADMET Prediction – PK Prediction framework, or simply, a M.A.P. workflow. Ultimately, using accurate and powerful in silico predictions can save you time and money by streamlining your discovery process.
Molecular Discovery: Designing with the End in Mind
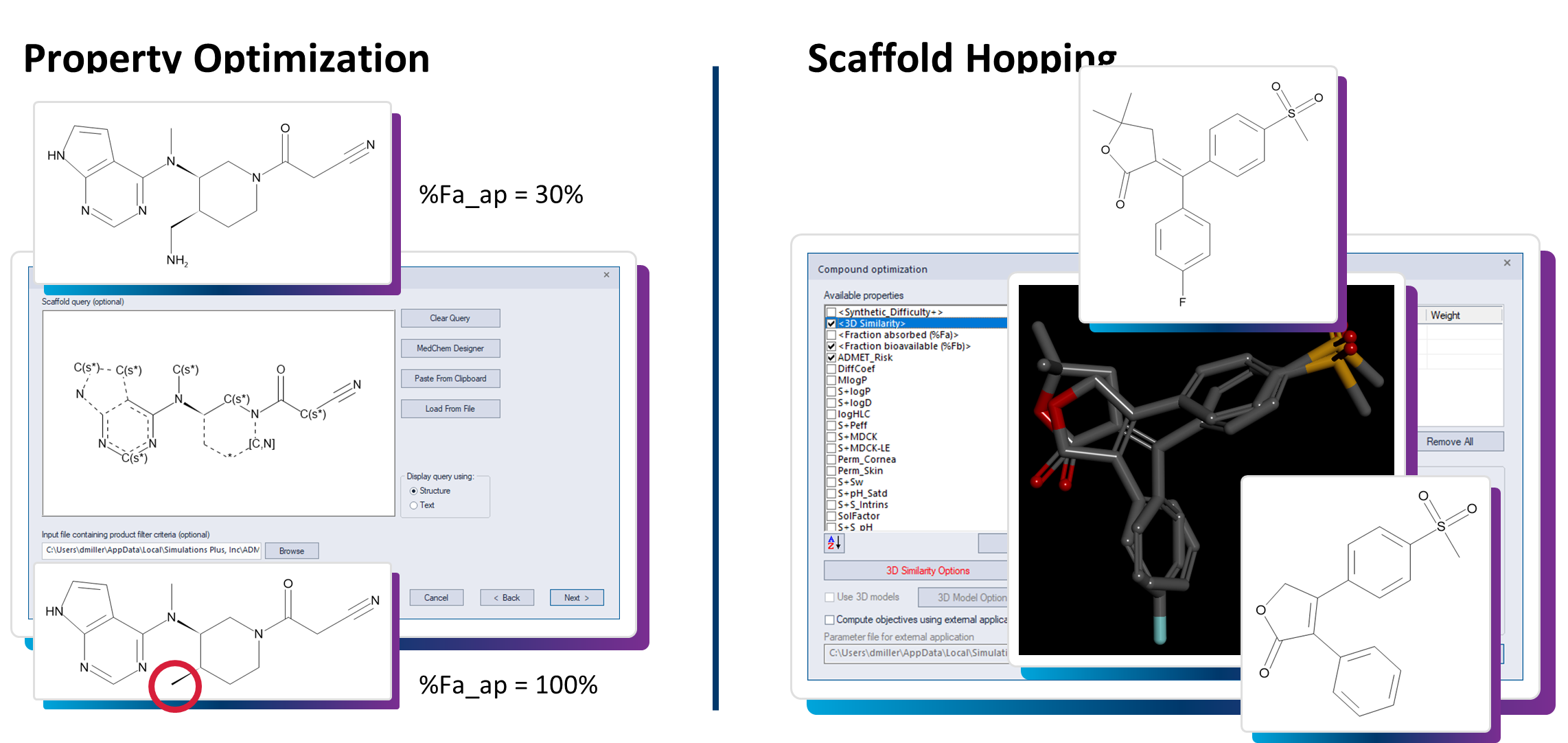
Modern molecular discovery is no longer limited to focusing on potency alone. By incorporating ADMET and PK insights into early design decisions, discovery chemists can pursue compounds that are more likely to succeed in vivo.
Several in silico platforms exist that offer cheminformatics solutions, with features ranging from scaffold clustering, R-group analysis, and multi-criteria decision analysis (MCDA) to support prioritizing compounds not just for activity, but for balanced profiles. Furthermore, the ability to generate 3D conformations to support conducting ligand-based virtual screening, or generative chemistry design using AI/ML algorithms such as AI-Driven Drug Design (AIDD), can facilitate researchers’ exploration of chemical space rapidly and effectively. Once compounds have been screened or designed, the process to triage results for physical synthesis and screening can be challenging without using quality predictions. Some platforms rely solely on volumetric shape and pharmacophore similarity, while others are based on QSAR models or electrostatics.
Molecular discovery efforts within ADMET Predictor® allows for chemists to design compounds with predicted ADMET properties, oral bioavailability or clearance potential in mind from the beginning. In turn, teams can therefore filter out candidates that might fail later due to poor absorption or rapid metabolism, ensuring every iteration moves the project closer to feasible candidates rather than theoretical ideals.
ADMET Prediction: Turning Data into Design Insight
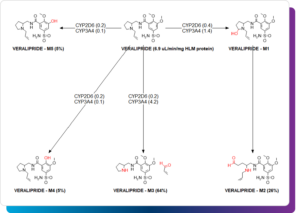
At the core of the M.A.P. workflow, is accurate, mechanistically informed ADMET prediction. While potency drives the initial discovery of compound classes, ADMET defines their survivability. Models that estimate properties such as pKa, solubility, permeability, metabolic stability, and toxicological endpoints allow teams to anticipate experimental outcomes before synthesis.
For example, predicting CYP-mediated metabolism, hERG binding, or AMES mutagenicity can highlight liabilities that might otherwise emerge only after months of bench testing. Similarly, modeling microsomal clearance across species helps bridge the gap between early discovery and DMPK evaluation.
Global models — built from large, curated datasets — offer broad predictive power but integrating local or proprietary data can further improve relevance. Tools that allow researchers to retrain or extend global models using their internal datasets create a feedback loop between computational and experimental work. This iterative refinement turns ADMET prediction into a living, adaptive component of discovery rather than a static filter applied at the end.
PK Prediction: Evaluating the Future
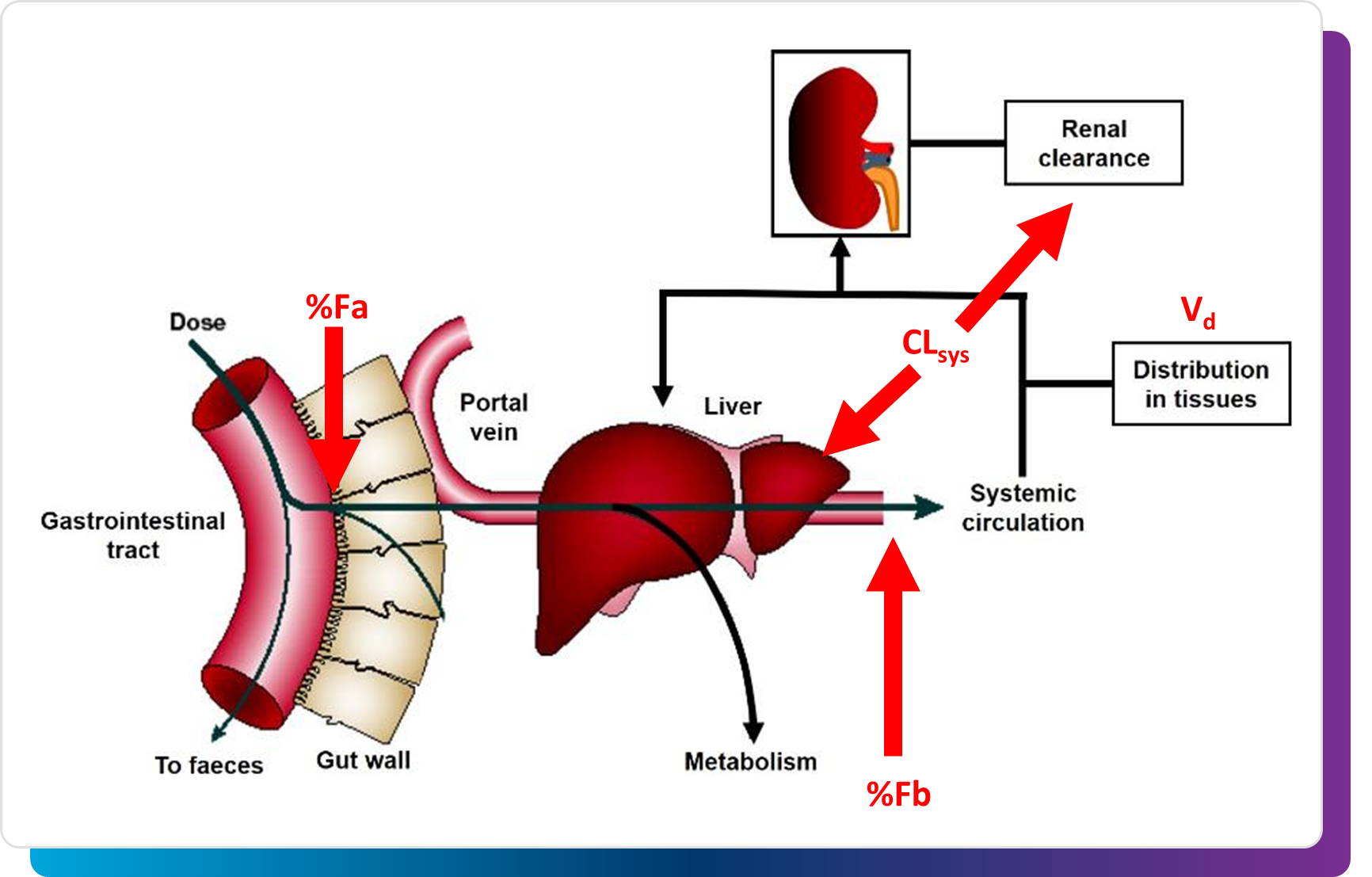
No matter how potent or safe a molecule is, its pharmacokinetic behavior ultimately determines whether it reaches its target in vivo. Traditionally, PK simulation has been reserved for later stages, though incorporating high-throughput PK simulations during the early discovery can provide essential context for prioritization.
Rapid predictions of key PK properties, such as fraction absorbed, systemic clearance, or volume of distribution can inform whether a compound is likely to achieve adequate exposure in animal models or human projections. Having this perspective early in the DMTA cycle allows teams to focus on molecules that are not only biologically interesting but also pharmacologically realistic.
This shift from reactive PK evaluation to proactive PK design helps align medicinal chemistry decisions with downstream development goals, saving time, money, and reducing experimental dead ends.
Integrating the M.A.P. Workflow
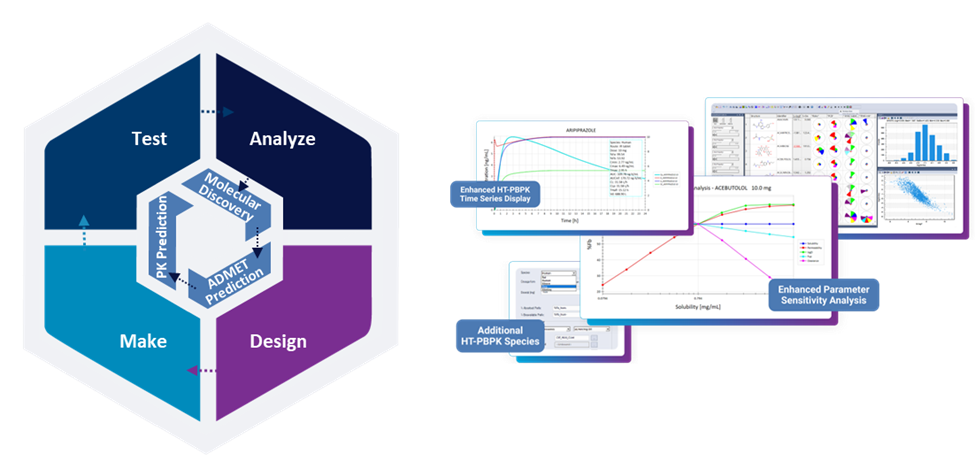
The full potential of in silico modeling emerges when these components — molecular design, ADMET prediction, and PK prediction — are connected. Integration across computational chemistry, medicinal chemistry, and DMPK functions ensures that every decision is informed by multiple perspectives.
Unfortunately, most discovery platforms only support individual functions or portions of the workflow. The only software that supports a broad workflow and practical implementation is ADMET Predictor®. The platform offers AI-driven drug design and ligand-based virtual screening capabilities alongside ADMET and PK prediction, which can be leveraged through the platforms’ GUI, via the REST API functionality, or through command line, Python and KNIME support.
This integration transforms the DMTA cycle from a linear sequence into a continuous feedback loop, where each iteration becomes smarter, faster, and more cost-efficient.
Building a More Predictive Future
The goal of early ADMET integration is not to replace experimentation but to make it more strategic — guiding synthesis toward compounds that are both promising and practical. By being “ADMET-aware” from the first iteration, discovery teams can reduce attrition, foster better collaboration, and bring a more predictive mindset to drug design.
To learn more about how integrated ADMET and PK modeling can enhance your discovery workflow, and explore the M.A.P. framework in depth, contact us to discuss how this approach might support your research goals.