The challenges of developing drug and biological products for rare diseases
A rare disease affects a small number of patients, and while quantitative definitions vary slightly across regions (Table 1), it is generally considered to affect fewer than 1 in 2,000 people when normalized by the regional population. While each specific rare disease impacts a relatively small group, the overall burden—considering the wide range of disease types and contributing factors—represents a significant area of unmet medical need (1).
- > 7000 types of rare disease
- ~ 300 million people live with rare diseases
- ~ 80% of rare diseases have a genetic cause
- ~70% of rare diseases with a genetic cause present in childhood
- ~ 95% lack approved treatments
- average time for an accurate diagnosis is 4.8 years
- ~ 30% of children with a rare disease die before age 5 years
Table 1: rare disease definitions by various agencies
| Region | Definition | Source |
| United States | < 200,000 [US population 334.9 million in 2023, approximately 1 in 1700] | The Orphan Drug Act (1983) |
| European Union | < 5 per 10,000 [equivalent to 1 in 2,000] | Regulation (EC) 141/2000 of the European Parliament |
| Japan | <50,000 [Japan population 123.7 million in 2024, approximately 1 in 2475] | The Article 77-2 of the Pharmaceutical Affairs Law |
| World Health Organization (WHO) | < 1 in 2000 | The landscape for rare diseases in 2024 (1) |
Developing a drug for a rare disease presents unique challenges due to the limited number of patients worldwide with any given condition. These challenges include, but are not limited to, the following (2, 3):
- Enrolling, engaging and retaining patients
- Designing and evaluating clinical trials
- Poor understanding of the disease process and natural history
- Incomplete understanding of clinically meaningful endpoints
- Inability to assess clinical benefit and achieve full approval
Nevertheless, the number of treatments for rare diseases approved by the Center for Drug Evaluation and Research (CDER) within the US Food and Drug Administration (FDA) has been growing. Figure 1 summarized the percentage of novel approvals by CDER/FDA and EMA from 2014 to 2024. During that period, FDA’s novel drug approvals for rare diseases exceeded 40% in 10 of those years and surpassed 50% in 5 years.
Other regulatory agencies have published similar statistics. For example, the number of medicines that have received an orphan designation from European Medicines Agency (EMA) between 2014-2024 was generally above 110 (4). In the year 2016, the number reached 209. However, the percentage of medicinal authorization with an orphan designation was lower compared to the % of CDER novel approvals for rare diseases (Figure 1). In Japan, a total of 432 orphan drugs were designated between 1993 fiscal year (FY) and 2018 FY, and 322 of them (75%) were approved (5).
Figure 1. Percentage of novel drug approvals (CDER /FDA) or medicinal authorization (EMA) for rare diseases (2014-2024)
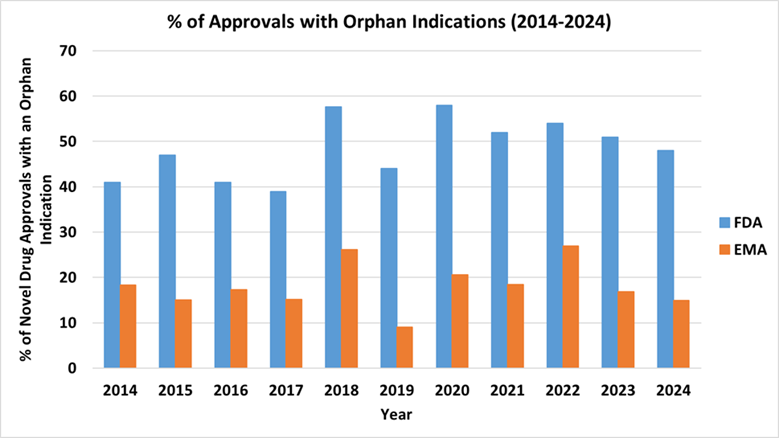
Source: generated based on FDA CDER Novel Drug Approvals reports from 2014 to 2024 (Novel Drug Approvals at FDA | FDA), and data published by EMA.
Clinical pharmacology and regulatory considerations for rare disease drug development
Clinical pharmacology plays a crucial role in any drug development program, including those for rare diseases. Clinical pharmacology considerations are specifically outlined in guidances entitled: “Rare Diseases: Early Drug Development and the Role of Pre-IND Meetings,” “Slowly Progressive, Low-Prevalence Rare Diseases With Substrate Deposition That Result From Single Enzyme Defects: Providing Evidence of Effectiveness for Replacement or Corrective Therapies,” and “Rare Diseases: Considerations for the Development of Drugs and Biological Products.”
Typically, clinical pharmacologists collaborate with multi-disciplinary product development teams to identify optimal dosage(s) for all potential patients. For drug programs targeting rare diseases, the same objectives must be met, but they are complicated by the unique challenge of having a limited patient population. As a result, model-informed drug development (MIDD) becomes particularly valuable in the context of rare disease drug programs. This is because MIDD has the potential to use sophisticated modeling and simulation techniques to combine and analyze data from multiple sources—such as clinical trials, preclinical studies, real-world evidence, and expert knowledge. By integrating all available knowledge, it improves our understanding of the disease, the drug’s mechanisms, and potential treatment outcomes. Furthermore, MIDD allows for the simulation of scenarios that have not yet been tested in clinical settings, such as varying patient populations, dosing regimens, or disease progression. This helps to inform decision-making and optimize the drug development process, even in the face of limited data and small patient groups. The role of MIDD is accentuated in the ‘N of 1’ trials which have been used in the development of individualized medicines to treat rare genetic diseases (6, 7). Ultimately, MIDD aids in making more accurate predictions about the drug’s efficacy, safety, and overall potential for success, which is especially important when studying rare diseases where traditional trial designs may not be feasible or cost-effective (8-10).
MIDD tools include but not limited to population pharmacokinetics (popPK), physiologically based pharmacokinetics (PBPK) and biopharmaceutics (PBBM), dose (exposure) – response (E-R) analysis, model-based meta-analysis, quantitative systems pharmacology (QSP) and toxicology (QST), agent-based models, disease progression models, and artificial intelligence / machine learning (11).
PopPK and E-R analysis have been routinely used to inform drug development and regulatory decisions. PopPK analysis helps identify and quantify sources of inter-individual variability (IIV) in drug’s PK, determines how factors such as age, body weight, renal/hepatic function, and genetics (e.g., CYP polymorphisms) influence drug exposure. PopPK in combination with E-R analysis and other modeling tools helps refine dosing recommendations for specific populations (e.g., pediatrics, elderly, renal/hepatic impairment). PopPK enables sparse sampling strategies, reducing patient burden while still generating meaningful PK data, which is particularly valuable in development programs for rare diseases. PopPK analyses were used in 100% of the CDER’s novel drug approvals treating rare diseases in 2024.
To learn how to perform PopPK and E-R analysis, explore available tools, services, and other resources at Simulations Plus.
PBPK/PBBM modeling is one type of MIDD tool. Its application in regulatory decision-making has been discussed and published previously (12, 13). Upon closer examination of its applications, over half of the novel drug approvals that involved PBPK analyses were designated as orphan drugs (Figure 2). PBPK/PBBM modeling has been used in regulatory submissions to evaluate the effect of intrinsic (such as age [pediatrics, geriatrics], enzyme / transporter polymorphism, and organ dysfunction [renal impairment, hepatic impairment]), and extrinsic (such as concomitant medications, food, and formulation) factors. More importantly, it can be used to evaluate the combination of several factors, which is challenging to assess in dedicated clinical trials especially for rare disease programs.
To learn how to perform PBPK/PBBM, explore available tools, services, and other resources at Simulations Plus.
Figure 2: Percentage of CDER novel drug approvals that contains PBPK modeling and simulation and with orphan indications.
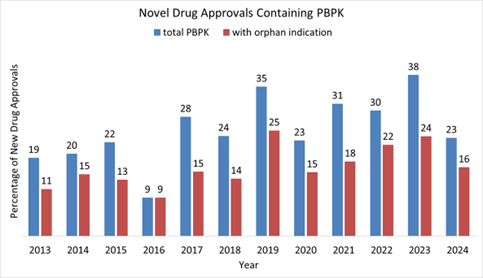
Source: Survey based on Novel Drug Approvals at FDA | FDA and Drugs@FDA
QSP is a modeling approach that can quantitatively and mechanistically integrate the mechanism of action of a drug to simulate patient responses to drug treatment with respect to efficacy and safety. The number of annual QSP model submissions to the FDA grew from single digits to over 80 between 2013 and 2023 (14). QSP modeling was applied to inform drug development across various therapeutic areas with cancer drug development programs accounting for 50% of total submissions (14). As of 2022, there were 121 submissions consisting of QSP modeling for informing rare disease drug development across development phases and therapeutic areas (15). Potential application of QSP in rare disease drug development include preclinical to clinical translation aiding in clinical trial design, dosage selection, informing safety assessment, extrapolation of efficacy/safety findings, integration of quantitative modeling of the natural history of a rare disease with QSP modeling, and to facilitate extrapolation and identification of biomarkers and clinical endpoints to quantitatively benchmark the progression of a rare disease (16).
To learn how to perform QSP modeling, explore available tools, services, and other resources at Simulations Plus.
Case Studies
In this section, case examples are provided for each type of MIDD tool.
PBPK
Trofinetide is indicated for the treatment of Rett syndrome, a rare genetic condition that affects brain development, in adults and pediatric patients 2 years of age and older. The recommended dosage is twice daily according to patient body weight and can be given with or without food. Trofinetide exhibits linear kinetics with no time- or dose-dependent effect on pharmacokinetic parameters. The effective elimination half-life of orally administered trofinetide in healthy subjects is about 1.5 hours. Trofinetide is not significantly metabolized by CYP450 enzymes. Hepatic metabolism is not a significant route of trofinetide elimination. Trofinetide is primarily excreted unchanged (approximately 80% of the dose) in urine, with minor excretion in feces, and not a substrate of a known kidney transporter. In vitro, trofinetide is a CYP3A4 inhibitor (17).
To evaluate the impact of renal impairment on the PK of trofinetide, a PBPK model was developed to guide dose selection in a phase 1 study (18). The PBPK model incorporated trofinetide physicochemical and biopharmaceutical properties and information on absorption, dissolution, and elimination that was determined experimentally or optimized during model development. Renal elimination of trofinetide was defined in the PBPK model by GFR multiplied by the fraction of unbound drug in the plasma (18). The model predicted that a 50% dose reduction is needed in subjects with moderate renal impairment to achieve similar exposure in healthy volunteers. The phase 1 study results confirmed the simulations results.
In addition, the PBPK model was used to evaluate the effect of trofinetide on the PK of a sensitive CYP3A substrate and ruled out clinically significant interaction with CYP3A substrates (19). Consequently, an unnecessary clinical DDI study was waived.
This example illustrates the best practice of utilizing PBPK modeling to address two different questions, impact of organ impairment and DDI liability. For organ impairment evaluations, PBPK modeling should be initiated early to enable broader patient enrollment in clinical trials without compromising safety.
PopPK and popPK/PD Analyses
Intravenous immunoglobulin (IVIG) is the only approved treatment for multifocal motor neuropathy (MMN), a rare, chronic, immune-mediated demyelinating neuropathy, with impaired grip strength being a common symptom, and a quantitative biomarker that is accepted as primary endpoint for this disease (20, 21).
A population PK modeling was applied to better understand the correlation between serum immunoglobulin G (IgG) levels with the improvement in grip strength in affected patients, specifically to characterize the interplay between dosing regimens and exposure of total IgG and the effects of patient factors on individual variabilities(22). The analysis demonstrated that optimizing the dosing regimen and dosing strategies using a precision dosing approach may play a key role in achieving therapeutic goals in patients with MMN. A subsequent model-based analysis characterized the dose–exposure–response analysis relationships in MMN, using grip strength as a clinical efficacy measure(23). A population pharmacokinetic–pharmacodynamic model was developed with data from a Phase 3 trial in patients with MMN. Simulations based on the model were then performed to investigate dose levels and dosing frequencies to achieve clinically meaningful improvements in grip strength (≥4 kg) in ≥70% of patients (23).
QSP
The complement cascade is part of the innate immune system, involved in eliminating pathogens through the generation of membrane attack complexes (MACs) that form pores in pathogen cell membranes. In normal conditions, complement activity can be initiated through the classical or lectin pathways and is amplified through the alternative pathway, resulting in assembly of MACs at the cell surface via the terminal pathway. This complex cascade includes a network of over 50 proteins with regulatory feedback and feedforward loops to ensure a measured pathogen response, while minimizing damage to healthy tissue.
Complement dysregulation is a key contributor to several rare diseases. These include paroxysmal nocturnal hemoglobinuria (PNH), atypical hemolytic uremic syndrome (aHUS), and C3 glomerulopathy (C3G) to name a few (24, 25). PNH is an acquired rare blood disorder affecting ~1 person per million (26), characterized by increased red blood cell death via complement system activation. Red blood cells affected by PNH are deficient in critical surface proteins, such as the complement regulators CD55 and CD59, making them susceptible to attack by the complement system. The anti-C5 monoclonal antibody eculizumab, effectively reduced intravascular hemolysis (27) . Regulatory approval of eculizumab for the treatment of PNH was revolutionary for PNH patients, provided proof-of-concept for complement-directed therapies, and provided reassurance that complement over-activity could be effectively reduced with manageable susceptibility to infectious diseases (27, 28). Despite the successes, an estimated 85% of eculizumab-treated PNH patients were found to have residual anemia (29) indicating persistent unmet medical need. Other complement-directed treatments have been approved for PNH treatment, improving the therapeutic options for patients (30) . Nonetheless, there are still opportunities to further develop complement-therapies directed at novel targets or better designed to inhibit complement without the requirement for high or frequent dosing (30) . Additionally, some complement-directed treatments have shown efficacy in other rare diseases where complement over-activity is implicated (e.g., aHUS, generalized myasthenia gravis, and neuromyelitis optica spectrum disorder). Given the landscape of complement-directed drug development, one can make a case for the benefit of modeling and simulation tools to consolidate our understanding of complement across diseases and facilitate the prediction of efficacy in novel targets or combination therapies.
QSP models of the complement system have been developed (31-35) to evaluate targets for efficacy in various therapeutic areas including PNH. For example, COMPLEMENTsym, a fluid-phase mechanistic QSP model of the complement alternative and terminal pathways includes a simulated population representing PNH patients. This population incorporates a representation of the underlying pathophysiology and is aligned with available clinical data for key analytes in PNH patients. In addition to a simulated population, representation of key approved therapies (e.g., eculizumab, pegcetacoplan, iptacopan) and their effects on both complement analyte concentrations and hemolysis, a key clinical endpoint for PNH, were built into the QSP model to provide reference points for future target evaluations. With a mechanistic model incorporating normal- and patho- physiology for complement, a PNH-specific simulated population, and representations of PNH standards of care, the QSP model is well situated to rapidly evaluate future treatments for PNH efficacy.
Further, the QSP model can be expanded to represent key target tissues, e.g., kidney, eye, to address tissue pathophysiology, whilst also accommodating inclusion of systemic biomarkers and endpoints. The ability of QSP models to be adapted to represent additional patient populations and relevant disease pathways offers opportunities for efficiently exploring novel rare disease treatments and bringing them to market sooner.
Conclusion
MIDD offers powerful tools that are revolutionizing the way we approach the treatment of rare diseases. By enabling more personalized and precise drug dosing, predicting drug interactions, and supporting regulatory approvals, MIDD approaches are helping to overcome the many challenges faced in rare disease drug development. As technology and data availability continue to improve, MIDD will undoubtedly be an indispensable part of the arsenal for clinicians and researchers working to develop effective treatments for these often overlooked and challenging conditions.
At Simulations Plus, we have a team of experts specializing in PBPK, PopPK and E-R analysis, QSP modeling, and medical communications. Don’t hesitate to get in touch if you need expert support or guidance—we’re here to help.
References:
- The Lancet Global H. The landscape for rare diseases in 2024. Lancet Glob Health. 2024;12(3):e341.
- Barrett JS, Betourne A, Walls RL, Lasater K, Russell S, Borens A, Rohatagi S, Roddy W. The future of rare disease drug development: the rare disease cures accelerator data analytics platform (RDCA-DAP). J Pharmacokinet Pharmacodyn. 2023;50(6):507-519.
- Pacanowski M, Vitarello J, Hyun I, Yu T, Zineh I. A Multistakeholder Perspective on Advancing Individualized Therapeutics. Clin Pharmacol Ther. 2023;114(5):994-1001.
- EMA. Orphan medicines in the EU. 2025 March 31, 2025. Available from: https://www.ema.europa.eu/en/documents/leaflet/infographic-orphan-medicines-eu_en.pdf.
- Sakushima K, Takeda H, Aoi Y. Orphan drug designation and development in Japan: 25 years of experience and assessment. Nat Rev Drug Discov. 2021;20(12):893-894.
- Kim-McManus O, Gleeson JG, Mignon L, Smith Fine A, Yan W, Nolen N, Demarest S, Berry-Kravis E, Finkel R, Leonard S, Finlayson S, Augustine E, Lyon GJ, Schule R, Yu T. A framework for N-of-1 trials of individualized gene-targeted therapies for genetic diseases. Nat Commun. 2024;15(1):9802.
- FDA. FDA In Brief: FDA Takes New Steps Aimed at Advancing Development of Individualized Medicines to Treat Genetic Diseases. 2025 Available from: https://www.fda.gov/news-events/press-announcements/fda-brief-fda-takes-new-steps-aimed-advancing-development-individualized-medicines-treat-genetic.
- Krishna R, Sebastien B, Corriol-Rohou S, Cheung SYA, Liu J, Suryawanshi S. Pediatric Rare Diseases Development in the Pharmaceutical Industry: An International Consortium for Innovation and Quality in Pharmaceutical Development, Clinical Pharmacology Leadership Group-Pediatrics Working Group, Rare Diseases Subteam Whitepaper Examining the Current Landscape and Recommendations for the Future. Clin Pharmacol Ther. 2024;116(6):1433-1441.
- Li RJ, Ma L, Li F, Li L, Bi Y, Yuan Y, Li Y, Xu Y, Zhang X, Liu J, Bhattaram VA, Wang J, Schuck R, Pacanowski M, Zhu H. Model-Informed Approach Supporting Drug Development and Regulatory Evaluation for Rare Diseases. J Clin Pharmacol. 2022;62 Suppl 2:S27-S37.
- Mitra A, Tania N, Ahmed MA, Rayad N, Krishna R, Albusaysi S, Bakhaidar R, Shang E, Burian M, Martin-Pozo M, Younis IR. New Horizons of Model Informed Drug Development in Rare Diseases Drug Development. Clin Pharmacol Ther. 2024;116(6):1398-1411.
- ICH. INTERNATIONAL COUNCIL FOR HARMONISATION OF TECHNICAL REQUIREMENTS FOR PHARMACEUTICALS FOR HUMAN USE (ICH) HARMONISED GUIDELIN: GENERAL PRINCIPLES FOR MODEL-INFORMED DRUG DEVELOPMENT M15. 2025 Available from: https://www.fda.gov/media/184747/download.
- Grimstein M, Yang Y, Zhang X, Grillo J, Huang SM, Zineh I, Wang Y. Physiologically Based Pharmacokinetic Modeling in Regulatory Science: An Update From the U.S. Food and Drug Administration’s Office of Clinical Pharmacology. J Pharm Sci. 2019;108(1):21-25.
- Zhang X, Yang Y, Grimstein M, Fan J, Grillo JA, Huang SM, Zhu H, Wang Y. Application of PBPK Modeling and Simulation for Regulatory Decision Making and Its Impact on US Prescribing Information: An Update on the 2018-2019 Submissions to the US FDA’s Office of Clinical Pharmacology. J Clin Pharmacol. 2020;60 Suppl 1:S160-S178.
- Bai JPF, Liu G, Zhao M, Wang J, Xiong Y, Truong T, Earp JC, Yang Y, Liu J, Zhu H, Burckart GJ. Landscape of regulatory quantitative systems pharmacology submissions to the U.S. Food and Drug Administration: An update report. CPT Pharmacometrics Syst Pharmacol. 2024;13(12):2102-2110.
- Bai JP, Wang J, Zhang Y, Wang L, Jiang X. Quantitative Systems Pharmacology for Rare Disease Drug Development. J Pharm Sci. 2023;112(9):2313-2320.
- Bai JPF, Stinchcomb AL, Wang J, Earp J, Stern S, Schuck RN. Creating a Roadmap to Quantitative Systems Pharmacology-Informed Rare Disease Drug Development: A Workshop Report. Clin Pharmacol Ther. 2024;115(2):201-205.
- FDA. NDA 217026 trofinetide oral solution US Prescribing Information. 2025 March 30, 2025. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2024/217026s001s003lbl.pdf.
- Darwish M, Marbury TC, Nunez R, Youakim JM, An D, Darling I, Lukacova V, Bishop KM. Physiologically-Based Pharmacokinetic Modeling of Trofinetide in Moderate Renal Impairment for Phase 1 Clinical Study Dose Selection with Model Validation. Eur J Drug Metab Pharmacokinet. 2025;50(1):23-38.
- FDA. NDA 217026 Trofinetide Clinical Pharmacology Review(s). 2025 March 30, 2025. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2023/217026Orig1s000ClinPharmR.pdf.
- FDA. BLA 89844 HYQVIA [Immune Globulin Infusion 10% (Human) with Recombinant Human Hyaluronidase] Solution, for subcutaneous administration PRESCRIBING INFORMATION. 2025 March 31, 2025. Available from: https://www.fda.gov/media/89844/download?attachment.
- FDA. BLA 70812 GAMMAGARD LIQUID, Immune Globulin Infusion (Human), 10% Solution, for intravenous and subcutaneous administration PRESCRIBING INFORMATION. 2025 March 31, 2025. Available from: https://www.fda.gov/media/70812/download.
- Li Z, Roepcke S, Franke R, Yel L. Dose, exposure, and treatment regimen of intravenous immunoglobulin G in multifocal motor neuropathy. Front Neurol. 2024;15:1478419.
- Li Z, Roepcke S, Franke R, Yel L. Dose-exposure-efficacy response of intravenous immunoglobulin G 10% in multifocal motor neuropathy. Ann Clin Transl Neurol. 2024;11(8):1977-1987.
- Reis ES, Mastellos DC, Yancopoulou D, Risitano AM, Ricklin D, Lambris JD. Applying complement therapeutics to rare diseases. Clin Immunol. 2015;161(2):225-240.
- Wong EKS, Kavanagh D. Diseases of complement dysregulation-an overview. Semin Immunopathol. 2018;40(1):49-64.
- NORD. National Organization for Rare Disorders: Paroxysmal Nocturnal Hemoglobinuria Available from: https://rarediseases.org/rare-diseases/paroxysmal-nocturnal-hemoglobinuria/.
- Hillmen P, Young NS, Schubert J, Brodsky RA, Socie G, Muus P, Roth A, Szer J, Elebute MO, Nakamura R, Browne P, Risitano AM, Hill A, Schrezenmeier H, Fu CL, Maciejewski J, Rollins SA, Mojcik CF, Rother RP, Luzzatto L. The complement inhibitor eculizumab in paroxysmal nocturnal hemoglobinuria. N Engl J Med. 2006;355(12):1233-1243.
- Dmytrijuk A, Robie-Suh K, Cohen MH, Rieves D, Weiss K, Pazdur R. FDA report: eculizumab (Soliris) for the treatment of patients with paroxysmal nocturnal hemoglobinuria. Oncologist. 2008;13(9):993-1000.
- Risitano AM, Peffault de Latour R. How we(‘ll) treat paroxysmal nocturnal haemoglobinuria: diving into the future. Br J Haematol. 2022;196(2):288-303.
- Ricklin D. Complement-targeted therapeutics: Are we there yet, or just getting started? Eur J Immunol. 2024;54(12):e2350816.
- Shoda L. Targeting Complement to Reduce Disease Activity: Quantitative Systems Pharmacology (QSP) to Enhance Efficient Drug Development: Part One. 2025 Available from: https://www.simulations-plus.com/resource/targeting-complement-to-reduce-disease-activity-quantitative-systems-pharmacology-qsp-to-enhance-efficient-drug-development-part-one/.
- Bakshi S, Cunningham F, Nichols EM, Biedzka-Sarek M, Neisen J, Petit-Frere S, Bessant C, Bansal L, Peletier LA, Zamuner S, van der Graaf PH. Mathematical Modelling of Alternative Pathway of Complement System. Bull Math Biol. 2020;82(2):33.
- Bansal L, Nichols EM, Howsmon DP, Neisen J, Bessant CM, Cunningham F, Petit-Frere S, Ludbrook S, Damian V. Mathematical Modeling of Complement Pathway Dynamics for Target Validation and Selection of Drug Modalities for Complement Therapies. Front Pharmacol. 2022;13:855743.
- Caruso A, Vollmer J, Machacek M, Kortvely E. Modeling the activation of the alternative complement pathway and its effects on hemolysis in health and disease. PLoS Comput Biol. 2020;16(10):e1008139.
- Zewde NT, Hsu RV, Morikis D, Palermo G. Systems Biology Modeling of the Complement System Under Immune Susceptible Pathogens. Front Phys. 2021;9.