
The goal of early drug discovery is to find novel lead compounds that have the desired potency, selectivity, and ADMET properties for pre-clinical evaluation. The figure below describes a typical lead discovery process. Shapes with a green background indicate areas where computational tools can have an impact.
01. discover
Computational tools for Early Lead Discovery
The process typically begins by testing a screening collection in the primary assay to find “hits”.
Two types of screening collections are shown; “general screening” and “targeted library”. This is the first area where computational tools can be incorporated. Compounds in small molecule drug discovery process should be reasonably soluble, have low (<5) logP value, good permeability and shouldn’t contain non-drug like fragments e.g. aromatic nitro groups. ADMET Predictor® can quickly and accurately predict solubility, logP, and permeability. These and other properties are incorporated into our ADMET Risk™ score. ADMET Predictor also contains an advanced query language so that one can define, save, and execute appropriate property thresholds. One can create separate query files suitable for drug, lead, or fragment-like libraries.
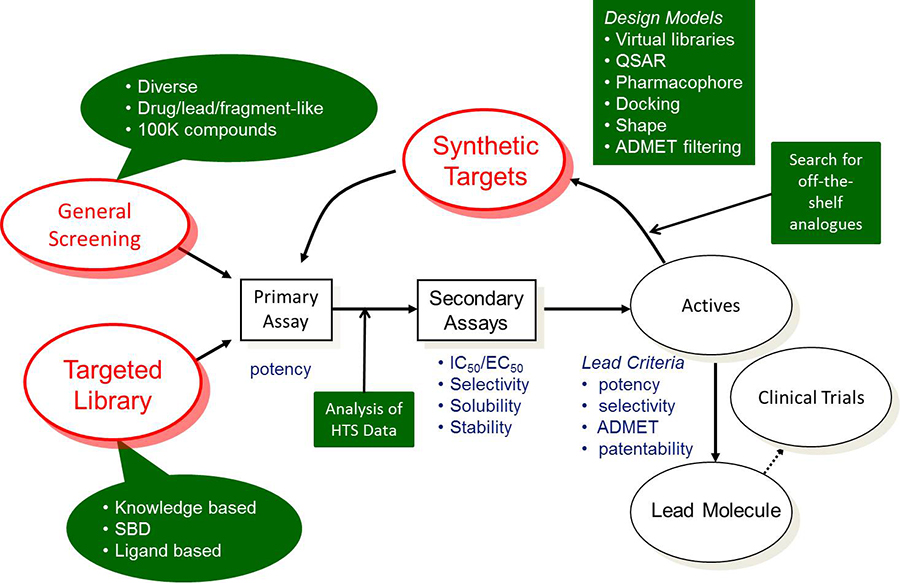
If too many compounds meet the criteria then one can select a diverse set using a tool based on k-means clustering of MDL MACCS fingerprints in the MedChem Studio module. Alternatively, one may want to generate a ligand based targeted library by finding compounds similar to known modulators of the same target. Here, one can use the similarity screening feature based on generation of molecular pairs. This approach offers several advantages over traditional fingerprint approaches, including the ability to align similar compounds by shared substructure and highlight their structural differences.
Analyzing Results
Analyzing the results of the high throughput screening campaign is a crucial step in the lead discovery process. Here, one can perform class generation to create clusters of compounds containing the same scaffold. The activity data in each cluster can be visualized with distribution plots to examine the clusters containing active compounds.
This also allows one to find anomalies. For example, a single compound in a class that has extremely high or low activity relative to other compounds in that class. These compounds need additional analysis, e.g. does the sample contain impurities, is the structure correct, etc.? Next, hit compounds are tested in secondary assays. One can also search for compounds that are similar to hit compounds. As data begins to accumulate, one can start to build a QSAR model using ADMET Modeler.
Medicinal chemists will analyze the hit compounds and develop synthetic schemes to produce analogues. Here, the combinatorial chemistry library enumeration tool in the MedChem Studio Module can be used to create a virtual library. Again, one would want to filter the library for acceptable ADMET properties. This will help to prioritize the list of synthetic candidates.
One can also use our combination of QSAR models with the GastroPlus® PBPK modeling platform to predict in vivo bioavailability. For example, one can simulate various doses, e.g. 1, 10, 100, or 1,000 mg, and predict fraction absorbed and oral bioavailability. This can also be used to prioritize synthesis of new compounds. After several loops through the process, one should identify several chemotypes with the desired potency, selectivity, ADMET and PK properties.