
The HTPK (High-throughput pharmacokinetic) Simulation Module is the preferred platform for discovery PBPK simulations and integrates the industry’s #1-ranked mechanistic absorption/PBPK models with #1-ranked machine learning ADME property predictions to rapidly predict all key in vivo endpoints in rats or humans, including:
01. Discover
What is the HTPK Simulation Module?
- Fraction absorbed (%Fa)
- Oral bioavailability (%Fb)
- Volume of distribution (Vd)
- Maximum plasma concentration (Cmax)
- Time at which maximum plasma concentration is reached (Tmax)
- Area under the concentration-time curve (AUC)
- In vivo half-life in rat and human (T1/2)
-
Calculation and optimization
It is also possible for optimal dose estimates to be quickly estimated within the virtual rat or human PBPK models and factor into compound prioritization decisions at the discovery stage. All calculation and optimization routines have been engineered for maximum computing performance, with capabilities to perform time-dependent mechanistic PK simulations at the rate of over 100 compounds per second!
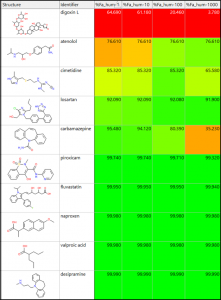
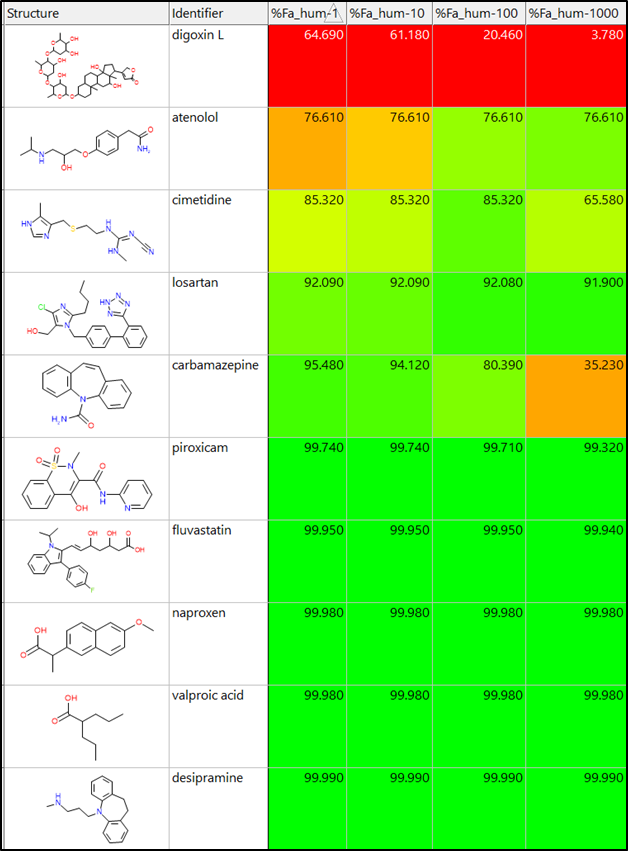
The table on the left shows %Fa predictions for ten drugs at 1, 10, 100, and 1000 mg doses. The spreadsheet is sorted in increasing order based on %Fa for a 1 mg dose.
Half of the drugs have a high fraction absorbed (>99%). Atenolol and losartan have lower %Fa values.
Aside from digoxin L, these two compounds have the lowest predicted human jejunal permeability (S+Peff) that limits their absorption. The fraction absorbed values for digoxin L, cimetidine, and carbamazepine decrease with increasing dose.
These compounds have lower predicted solubility at pH 7.4 than the other compounds in the data set.
-
The algorithm uses our top-rated Advanced Compartmental Absorption and Transit (ACAT™) model
The algorithm uses our top-rated Advanced Compartmental Absorption and Transit (ACAT™) model from GastroPlus to simulate dissolution, transit, and absorption in the GI tract. The rapid dose optimization capabilities provide flexible options to determine the estimated dose (D) needed to reach different types of user-specified concentrations:
- Average plasma concentration (Cave)
- Minimum plasma concentration (Cmin)
- Maximum plasma concentration (Cmax)
Predictions using the HTPK Simulation Module can be generated from structural information alone. Workflows and templates are also available to intuitively integrate your experimental values into the simulation routines, if available, to increase the accuracy of the predictions. Advanced setup capabilities allow the combined use of experimental (preferred) and calculated (fallback) values within the same run.
The module can be easily run at the command line to incorporate it into KNIME, PipelinePilot™, or other programs such as SpotFire®.
-
Examples of HTPK Simulation Output Include:
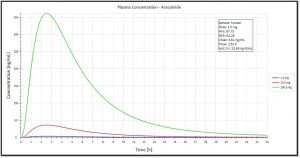
Display of CPU time at different concentrations:
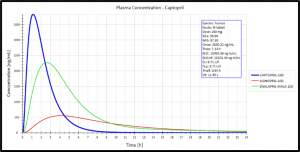
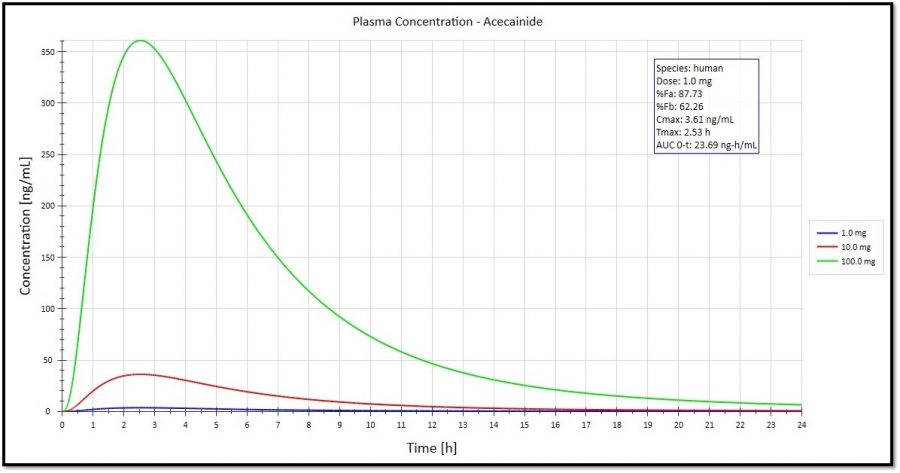
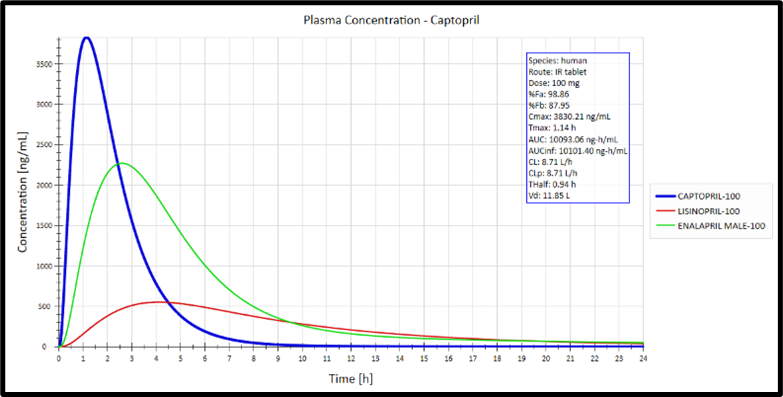
Overlay of Cp-time curves for different compounds:
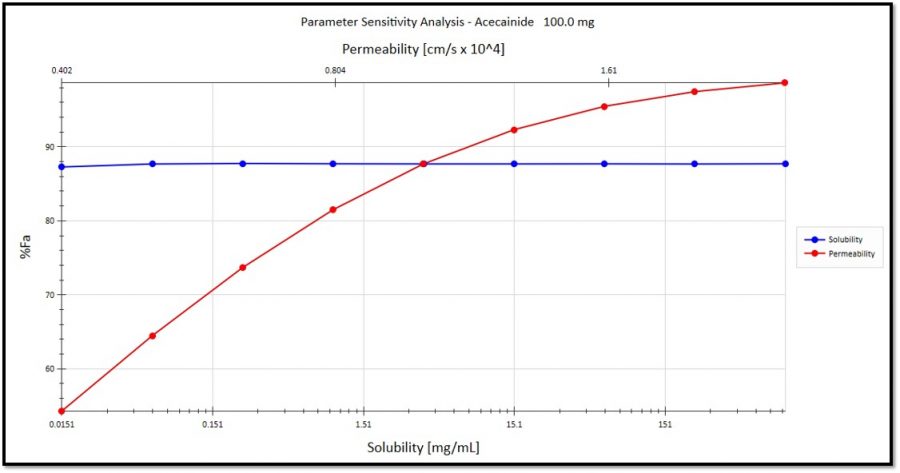
Parameter sensitivity analysis %Fa vs. solubility and permeability:

02. What's New

R&D collaborations with our large pharmaceutical partner established the functionality requirements in the HTPK Simulation Module necessary to bridge communication between discovery and early preclinical/DMPK development, and the new options for implementation provide the flexibility needed to fit almost any company’s internal workflow processes:
- New Models: New models offer increased accuracy and granularity of your predictions, including areas such as liver microsomal and hepatocyte intrinsic clearance for monkeys and dogs, plasma protein binding (Fup) for monkeys and dogs, melting point, Gibbs free energy of self-solvation and hydration, and more.
- Enhanced Models: Updates to our trusted models offer improved understanding around aqueous solubility, volume of distribution, blood-brain barrier (BBB) penetration, and more.
- Updated Modules: The HTPK and AIDD modules have been expanded to provide more detail to your predictions and promote novelty and synthetic feasibility.
- Expanded REST API: The latest version of our REST API supports image generation in Linux, easier property prioritization, and more.
- + more
03. Resources
Explore Resources
Join your peers worldwide to explore the discovery PBPK opportunities within ADMET Predictor