Effective de novo drug design is a long-standing but elusive goal for computational chemistry. Artificial intelligence (AI) and machine learning (ML) technologies have ushered in a new era in computer aided drug design (CADD), and there is huge potential to leverage technology to cure humankind’s cruelest diseases. However, AI isn’t fairy dust, and sprinkling it over every challenge won’t automatically produce solutions. As with any new technology, it is the careful and reasoned application of it that will lead to success.
AI/ML-powered CADD can accelerate drug identification and optimization. The rapid growth in computing power, data availability, and advanced algorithms have led to breakthroughs in every stage of the drug development process. These advances can reduce development costs and provide safer, more effective therapies for patients. This is important because even now, nearly 90% of compounds entering clinical trials fail to reach approval.
The last few years have seen an explosion of companies using AI/ML techniques to improve certain aspects of the drug development process, but much of this effort has focused on designing molecules with better target affinity and activity—even though these issues are rarely the cause of clinical failure1. The reality is that most of the candidates that fail do so because of unmanageable toxicity or poor drug-like properties. Thus, to have any real impact, de novo drug design platforms must consider absorption, distribution, metabolism, excretion, and toxicity (ADMET) as well as pharmacokinetic (PK) properties in the earliest stages of the design process.
How Do I Know If an AI Solution Will Provide Reliable Predictions?
The key to any in silico prediction—from using multi-omics data to predict new targets to using advanced physiologically-based pharmacokinetic (PBPK) models to predict tissue distribution of a drug in specific subsets of patients—is data.
While the choice of AI/ML approach to be used is extremely important and must be carefully selected for the specific goal, those algorithms must then be trained on a sufficiently large and carefully curated data set. This requires a nuanced understanding of the data, including the biology of the target and the details of the assays used to acquire the data. If the data used to build/train the algorithm isn’t large enough or of high enough quality, then it won’t matter what approach you choose; your predictions won’t be any good. If you’re looking to speed up your drug discovery process, what you really need are AI/ML solutions that have been refined over time by scientists who understand the biology behind the data to ensure that the algorithms are meaningful.
Let’s take a look at some examples where understanding the biology behind the data is essential for to creating robust and accurate models.
Let’s say you want to develop novel small molecule toll-like receptor 2 (TLR2) antagonists to reduce inflammation and to do so, you would like to build a QSAR model using known TLR2 antagonists. While the biology is complicated, it’s important to at least know that TLR2 is a membrane protein, a receptor which is expressed on the surface of certain cells and recognizes foreign substances causing the activation of NF-kB, typically leading to the secretion of inflammatory cytokines (Fig 1A). It’s also important to know that it forms a heterodimer with TLR1 or TLR6, and which of these heterodimers are formed as well as which ligand is used to stimulate TLR2 will have different effects on its activity. This knowledge becomes essential as you begin your search for data with which to build your QSAR model.
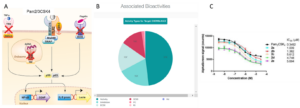
Figure 1:The Difficulties with Data. A) HEK-Blue TLR2 activity assay (from https://www.invivogen.com/hek-blue-lucia-htlr2). TLR2 is expressed in HEK cells with either TLR1 or TLR6. Activation by Pam3CSK4 or Pam2CSK4 respectively causes intracellular signaling resulting in the activation of NF-kB transcription factor (p50/p65). This then induces the transcription of the SEAP reporter gene, which is easily quantified with the SEAP blue assay. TLR2 inhibitors can be identified by incubating the cells with candidate molecules and quantifying the decrease in reporter activity. B) Chart of bioactivities of compounds targeting TLR2 in ChEMBL (TLR2 is Target CHEMBL4163), broken down by activity type. C) Data from Arai et al, which is listed as “IC50” activity type in ChEMBL, but is actually competitive displacement, not inhibition.
Common starting points for building a dataset are academic literature, patents, ChEMBL2, and of course your own lab data should you have any.
Looking at the literature, several academic and industry research groups have published results in which they synthesized and tested small molecules for their ability to inhibit TLR2 signaling 3–8. In total, this includes well over 100 structures with associated TLR2 inhibition data. While the publication from Neuropore did not disclose any structures, a patent search reveals data on ~50 compounds from this group9. Finally, a search for “TLR2” as a target in ChEMBL returns data on over 800 compounds, including 239 listed as “Inhibition” or “IC50” activity types (Fig 1B). The initial inclination is to download all of this data, combine it, and use it for training a QSAR model; however, a closer inspection of the data demonstrates why this is a strategy that’s doomed to fail. There are several questions you must ask when building a dataset:
- Are the assays used to quantify the inhibition comparable? The ideal scenario is that all of the data comes from one lab using the same assay. But outside of big pharma, that’s rarely the case. In lieu of such datasets, we must carefully consider that data sources to determine if we can combine them.
Let’s compare data from Mistry et al6 and Xu et al5. Xu et al use the now standard HEK-Blue SEAP assay to quantify inhibition (Fig 1A). Briefly, cells that express TLR2/1 or TLR2/6 are stimulated with Pam3CSK4 (Pam3CysSerLys4, a synthetic triacylated lipopeptide) or Pam2CSK4 respectively10. This activates NF-kB, which in turn induces the expression of secreted embryonic alkaline phosphatase (SEAP), which can be easily quantified in the cell culture media11. Mistry et al tested their compounds before this specific assay was developed, instead using an assay in which HEK cells were transiently transfected with TLR2/1 or TLR2/6 and stimulated with Pam3CSK4 or Pam2CSK4, then quantifying activity by the amount of IL-8 mRNA produced via RT-qPCR.
Because the techniques used to quantify activity are so different, it isn’t appropriate to combine these data for model training, unless there exists some direct comparison between them. In this case, we are lucky. Although Habas et al did not reveal any structures in their publication, they did use the best compound (C29) from Mistry et al as a control in their studies, and they used the HEK-Blue SEAP assay7. The IC50 of C29 was found to be 57.6uM in the SEAP assay while it was reported to be 37.6uM in the RT-qPCR assay. That the values were in the same ballpark gives us confidence that the two datasets could be integrated, with a possible scaling factor of 57.6/37.6 for the Mistry et al data.
Another group developed TLR2 inhibitors and used the HEK-Blue SEAP assay, but they reported their results as percent inhibition at 25uM8. While this data cannot be integrated with the other data to train a regression model, it could be used to train a classification model, which can be powerful in its own right.
This approach offers several advantages. First, it increases the size and diversity on which your model is trained by including additional data for which IC50 values are not available. Second, the classification model can be used as a determining descriptor in the regression model; in other words, a compound would have to pass the classification model (i.e., be predicted to be an inhibitor) to have its IC50 predicted by the regression model. This provides increased odds of arriving at a real, potent TLR2 inhibitor.
- Is the assay really an inhibition assay, or is it simply a binding/displacement assay labeled as such? Many research groups will report as a compound’s “IC50” the concentration at which it can displace a known ligand from binding to a target. Take for example the data from Arai et al, which is described in the ChEMBL dataset (Fig 1B) as “IC50” activity type12. The researchers “performed a competitive binding assay based on the Alpha Screen system using TLR2-conjugated acceptor beads, streptavidin-coated donor beads, and Pam2CSK4-biotin as a tracer… competitive ligands competed with Pam2CSK4-biotin, which decreased the signal.” Such an assay does not tell you if a compound activates or inhibits the receptor; it only tells you if the ligand binds. If you were to just look at the data presented in ChEMBL and the paper (Fig 1C), it would lead you to believe that these compounds are indeed inhibitors and worthy of being included in your training dataset, but the authors of the paper go on to show that these compounds are actually agonists! Including these compounds in your TLR2 inhibitor data for training would completely ruin your model.
- Is the compound actually a ligand of the target? In other words, beware the phenotypic assay. Zhou et al reported the discovery of a novel “TLR2 signaling inhibitor” which was identified using “a sensitive cell line stably expressing TLR2, CD14, and an NF-κB-driven-luciferase reporter gene” stimulated not with Pam3CSK4 but instead with lymphocytic choriomeningitis virus13. While the reported compound inhibited the luciferase signal, the researchers did not determine if the compound actually interacts with TLR2. There are no competitive binding studies, no SAR studies, and no comparisons to known TLR2 inhibitors. While the compound may indeed be a TLR2 ligand, it would be better to error on the side of caution and exclude this compound from the combined dataset.
- Are the compounds binding to the same pocket? If not, they shouldn’t be combined to train a model. The compounds from Neuropore are actually allosteric binders, so even though a comparable assay is used, they cannot be combined with the other compounds that bind to the active site. You may want to use these compounds to design novel allosteric inhibitors, but that would require a model distinct from that used to design competitive inhibitors.
The examples cited here are specific to designing a QSAR model, but they are generally applicable to developing a predictive model for any property. For instance, many of the same assay variability issues arose as we were building our new cytochrome P450 (CYP) activity models. While there is a great deal of data on the activity of compounds toward various CYPs in the literature and in ChEMBL, much of this data that is labeled “IC50” is actually from competitive binding assays and does not measure biologic inhibition. Careful analysis of the data was necessary to build appropriate datasets for model training.
Likewise, knowledge about the differences in assays is very important. For instance, when developing models to predict permeability, there are several different assays that are commonly used, including PAMPA, Caco-2 cells, and various versions of MDCK cells, and often significant lab-to-lab variability even when using the same assay. Models have to be built separately for each assay, or carefully scaled using identical control compounds tested in each assay.
In silico predictions are only as good as the data from which they are built. This is where many AI and machine learning solutions can’t hit the mark—they utilize the core technology but don’t have the biologic expertise it requires to create robust and accurate models that can account for the complexities of the human body and disease states.
Open-source options for ADMET/PK data in the public domain are often employed, but they lack both the quantity and quality of data needed for accurate model building, regardless of the AI/ML methodology used.
Simulations Plus (SLP) has been a leader in the field of small molecule ADMET and PK property prediction for over two decades, with our flagship ADMET Predictor® and GastroPlus® software being used and consistently validated by more than 100 biotech and pharma companies14–17. We have leveraged these relationships to access a treasure trove of industry ADMET/PK data in addition to all publicly available data to build our models. For example, our ionization constant model (pKa)18 is built from the largest dataset in the world, now with over 47,000 compounds from academic and industry sources. We have also taken considerable time and effort to hand-curate our datasets to ensure that they are of the highest quality. We have built predictive models for all relevant ADMET/PK parameters using a variety of ML & mechanistic techniques, and we offer the ability to develop your own models from your own data using these techniques via our ADMET Modeler software. No other company has the capacity that we do to rapidly and accurately predict the parameters most relevant to clinical success – ADMET and PK properties.
Moreover, we recently released the AI-driven Drug Design (AIDD™) platform, which automates the drug design process by integrating our high-throughput PBPK simulations and ADMET predictions with our advanced generative chemistry algorithms19. AIDD performs iterative multi-objective optimization to evolve novel molecules that are active and lead-like. Again, we focused on quality and quantity in the de novo design process, in the ADMET/PK models that can be used in the multi-objective optimization process as well as in the way novel molecules are evolved from seed compounds and our extensive filters to ensure only drug-like compounds are produced. While other companies often pay lip service to including ADMET and PK property prediction in their de novo design platforms, no one else has truly focused on the integration of ML and mechanistic PBPK simulations in drug design to the same extent, and they don’t have the data to back it up. AIDD was designed by experienced medicinal chemists and developed in collaboration with industry partners to produce the most advanced integrated de novo drug design platform on the market.
How Do I Get Started Using AI in My Drug Discovery Program?
It is no exaggeration to say that the early drug discovery software and support biotechs select can make or break their ability to survive. Hopefully, this blog post has helped you identify key issues in developing AI/ML algorithms to predict important properties of novel molecules and made you think more carefully about what to consider when comparing de novo drug discovery platforms and consultants.
Speed and reliability of predictions are what make for a good solutions partner, and nobody on the market can match the Simulations Plus AIDD software platform or our Early Drug Discovery team.
If you’re interested in learning more about leveraging our software or experts in your drug discovery program, let us know.
References
- Sun D, Gao W, Hu H, Zhou S. Why 90% of clinical drug development fails and how to improve it? Acta Pharmaceutica Sinica B. 2022;12(7):3049-3062. doi:10.1016/j.apsb.2022.02.002
- Bento AP, Gaulton A, Hersey A, et al. The ChEMBL bioactivity database: an update. Nucleic Acids Res. 2014;42(Database issue):D1083-1090. doi:10.1093/nar/gkt1031
- Cheng K, Wang X, Zhang S, Yin H. Discovery of small molecule inhibitors of the TLR1-TLR2 complex. Angew Chem Int Ed Engl. 2012;51(49):12246-12249. doi:10.1002/anie.201204910
- Grabowski M, Murgueitio MS, Bermudez M, Rademann J, Wolber G, Weindl G. Identification of a pyrogallol derivative as a potent and selective human TLR2 antagonist by structure-based virtual screening. Biochem Pharmacol. 2018;154:148-160. doi:10.1016/j.bcp.2018.04.018
- Xu Q, Li T, Chen H, Kong J, Zhang L, Yin H. Design and optimisation of a small-molecule TLR2/4 antagonist for anti-tumour therapy. RSC Med Chem. 2021;12(10):1771-1779. doi:10.1039/D1MD00175B
- Mistry P, Laird MHW, Schwarz RS, et al. Inhibition of TLR2 signaling by small molecule inhibitors targeting a pocket within the TLR2 TIR domain. Proceedings of the National Academy of Sciences. 2015;112(17):5455-5460. doi:10.1073/pnas.1422576112
- Habas A, Reddy Natala S, Bowden-Verhoek JK, et al. NPT1220-312, a TLR2/TLR9 Small Molecule Antagonist, Inhibits Pro-Inflammatory Signaling, Cytokine Release, and NLRP3 Inflammasome Activation. Int J Inflam. 2022;2022:2337363. doi:10.1155/2022/2337363
- Cai S, Zhu G, Cen X, et al. Synthesis, structure-activity relationships and preliminary mechanism study of N-benzylideneaniline derivatives as potential TLR2 inhibitors. Bioorganic & Medicinal Chemistry. 2018;26(8):2041-2050. doi:10.1016/j.bmc.2018.03.001
- Wrasidlo WJ, Stocking E, Natala SR. Compounds and compositions as modulators of tlr signaling. Published online December 17, 2021. Accessed September 11, 2023. https://patents.google.com/patent/CR20210499A/en
- Brandt KJ, Fickentscher C, Kruithof EKO, Moerloose P de. TLR2 Ligands Induce NF-κB Activation from Endosomal Compartments of Human Monocytes. PLOS ONE. 2013;8(12):e80743. doi:10.1371/journal.pone.0080743
- HEK-BlueTM hTLR2-TLR1. InvivoGen. Published August 3, 2018. Accessed September 11, 2023. https://www.invivogen.com/hek-blue-htlr2tlr1
- Arai Y, Inuki S, Fujimoto Y. Site-specific effect of polar functional group-modification in lipids of TLR2 ligands for modulating the ligand immunostimulatory activity. Bioorg Med Chem Lett. 2018;28(9):1638-1641. doi:10.1016/j.bmcl.2018.03.042
- Zhou S, Cerny AM, Bowen G, Chan M, Kurt-Jones EA, Finberg RW. Discovery of a novel TLR2 signaling inhibitor with anti-viral activity. Antiviral Res. 2010;87(3):295-306. doi:10.1016/j.antiviral.2010.06.011
- Soliman ME, Adewumi AT, Akawa OB, et al. Simulation Models for Prediction of Bioavailability of Medicinal Drugs-the Interface Between Experiment and Computation. AAPS PharmSciTech. 2022;23(3):86. doi:10.1208/s12249-022-02229-5
- Sohlenius-Sternbeck AK, Terelius Y. Evaluation of ADMET Predictor in Early Discovery Drug Metabolism and Pharmacokinetics Project Work. Drug Metab Dispos. 2022;50(2):95-104. doi:10.1124/dmd.121.000552
- Choi SM, Kang CY, Lee BJ, Park JB. In Vitro-In Vivo Correlation Using In Silico Modeling of Physiological Properties, Metabolites, and Intestinal Metabolism. Curr Drug Metab. 2017;18(11):973-982. doi:10.2174/1389200218666171031124347
- Rukthong P, Sereesongsang N, Kulsirirat T, Boonnak N, Sathirakul K. In vitro investigation of metabolic fate of α-mangostin and gartanin via skin permeation by LC-MS/MS and in silico evaluation of the metabolites by ADMET predictorTM. BMC Complement Med Ther. 2020;20(1):359. doi:10.1186/s12906-020-03144-7
- Fraczkiewicz R, Lobell M, Göller AH, et al. Best of both worlds: combining pharma data and state of the art modeling technology to improve in Silico pKa prediction. J Chem Inf Model. 2015;55(2):389-397. doi:10.1021/ci500585w
- AIDD, an interactive AI-driven drug design system that uses molecular evolution and mechanistic pharmacokinetic simulation to optimize multiple property objectives simultaneously. doi:10.21203/rs.3.rs-3270269/v1