
ADMET Modeler is a QSAR/QSPR model building module in ADMET Predictor®. It automates the difficult and tedious process of making high quality predictive QSAR/QSPR models from experimental data sets. It works seamlessly with ADMET Predictor’s descriptors as its inputs and appends the selected final model back to ADMET Predictor as an additional predicted property.
01. Discover
What is ADMET Modeler™?

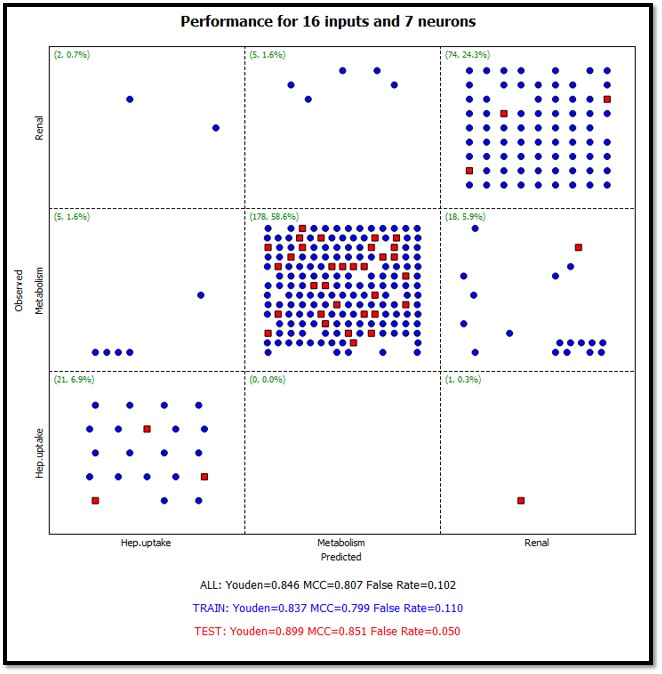
Multi-class (greater than 2) classification models. The diagram below shows the confusion matrix for a 3 class ANNE model.
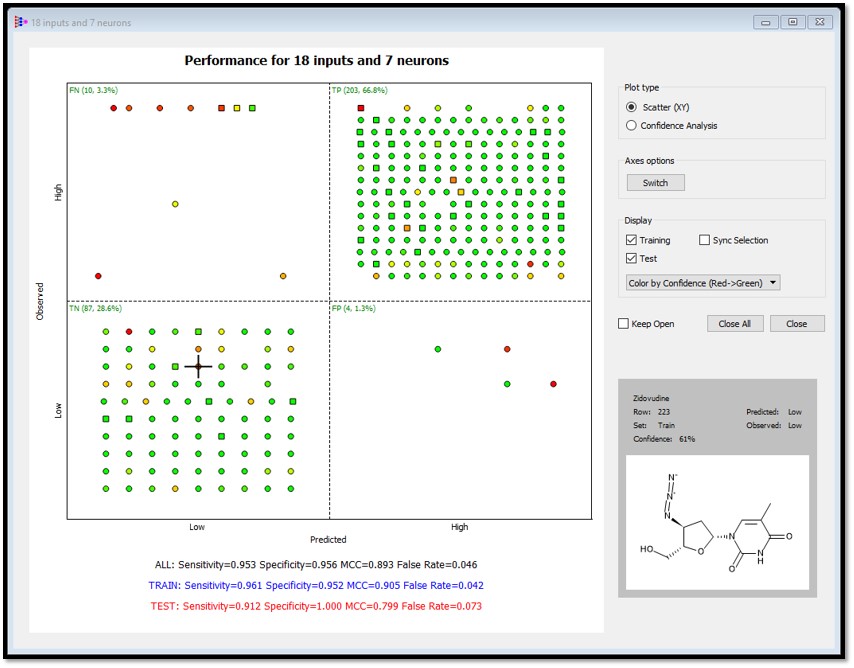
Performance plots can be colored by confidence. In the example below, confidence estimates are colored from red (low confidence) to green (high confidence). Also note that the confidence estimate for the compound is displayed in the lower right hand corner when a point in the plot is selected.
DELTA™ model approach is now automated
02. Explore
Modeling Algorithms provided in ADMET Modeler
-
The following modeling methods are offered in ADMET Modeler:
- Kohonen Self-Organizing Maps
- Artificial Neural Network Ensembles for regression and binary classification models
- Support Vector Machine Ensembles for regression and classification models
- Kernel Partial Least Squares and Ordinary PLS for regression
- Multiple Linear Regression
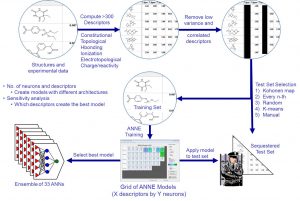
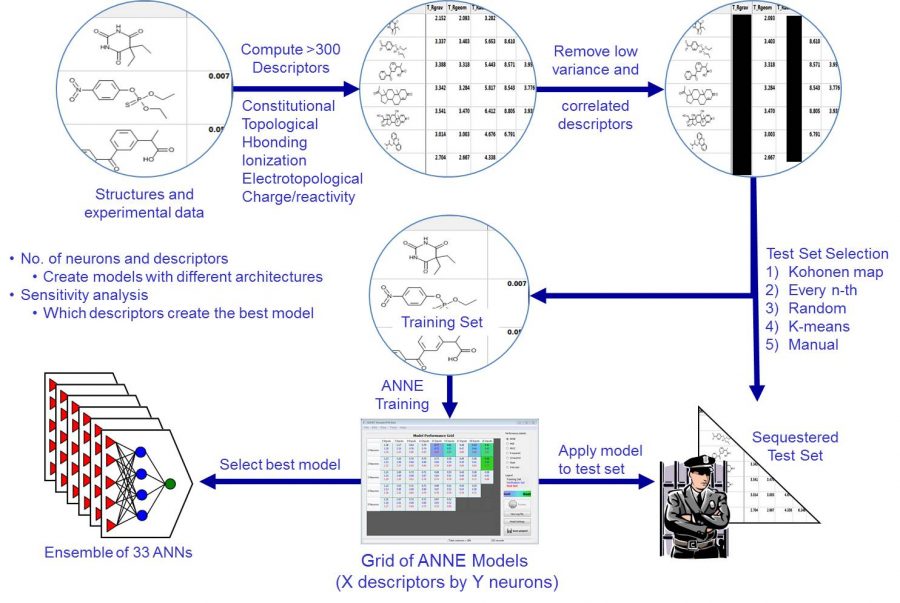
The first image on the left shows a typical workflow to create an artificial neural network model. ADMET Predictor contains over 300 atomic and molecular descriptors. The modeling algorithm automatically removes low variance and correlated descriptors. Several test set selection methods are available such as Kohonen maps and K-means. The test set is never used in model training, so it represents an external test set. The number of descriptors and neurons needed to create the best model is not known a priori. Thus, we create a grid of different architectures, i.e., different number of neurons and descriptors. There are also several algorithms to select the best model.
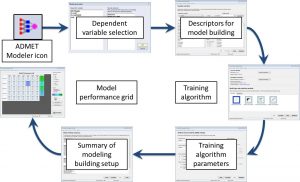
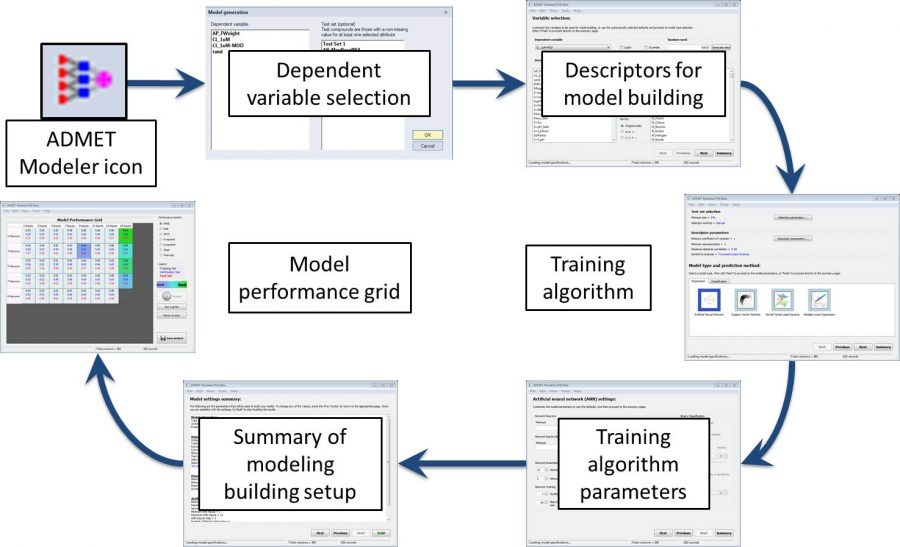
The above workflow has been incorporated into a model building wizard in ADMET Modeler. A diagram of the wizard is shown as the second image on the left.
-
Modeling runs became much, much faster . . .
When the original predictive models for physicochemical and biopharmaceutical properties for ADMET Predictor were developed using a generic modeling software, the time required to develop each model using all of the above steps was as much as 2-3 months. Now, the same models have been retrained with ADMET Modeler using the same steps, but with even more descriptors, in as little as a few hours! This dramatic productivity improvement is achieved through automation of all the tedious tasks that were previously done manually, as well as through highly efficient training code optimized for speed.
And the models are better . . .
The product of this new methodology is not only greater speed, but also better models. Now a wider set of architectures can be fitted rapidly, so more options can be investigated to see which is best. Selecting the best model from a larger set of models produces a final model that is more accurate, and usually smaller (fewer descriptors and parameters, in accordance with Occam’s Razor principle).
. . . and interpretable
Our new Descriptor Sensitivity Analysis tool removes the “black box” stigma usually affixed to Artificial Neural Network (ANN) models as well as their ensembles (ANNE). The ANN(E) response for a given compound is intuitively visualized via gradients calculated per each individual descriptor. Thus, the trends in predicted property can be easily tied to local changes in descriptors. After automatic identification of the most influential descriptors, a series of new chemical derivatives of the compound in question can be easily designed – derivatives that push the property of interest in the desired direction.

03. Resources
Explore Resources