ADMET Risk™
The original Rule of 5 is widely considered to be an important development in modern drug discovery (Lipinski, et al; 1997). The Rule of 5 takes on numeric values from 0 to 4 as a measure of the compounds potential absorption liability. As such, the Rule of 5 is a useful computational filter in drug candidate screening. In terms of ADMET Predictor descriptors and models, the Rule Of 5 model rules can be formulated as follow the following set of conditions:
- MlogP > 4.15 (excessive lipophilicity)
- MWt > 500 (large size)
- HBDH > 5 (too many potential hydrogen bond donors)
- M_NO > 10 (too many potential hydrogen bond acceptors)
Most commercial drugs suitable for oral dosing violate no more than one of the rules these conditions represent.
As an extension of that concept, Simulations Plus has created a series of “ADMET Risk” rule sets and calibrated them against our own ADMET models. They are parameterized to include thresholds for a wide range of calculated and predicted properties that represent potential obstacles to a compound being successfully developed as an orally bioavailable drug. These thresholds were obtained by focusing in on a specific subset of drugs in the World Drug Index (WDI). Similar to the methodology used by Lipinski et al., we removed irrelevant compounds from a 2008 version of the WDI. In particular, we removed phosphates, antiseptics, insecticides, emollients, laxatives, etc., as well as any compound that did not have an associated United States Adopted Name (USAN) or International Non-proprietary Name (INN) identifier. The structure of the principal component in salts was extracted and neutralized, after which duplicate structures were removed. This left us with a data set of 2,316 molecules, 8.3% of which violated more than one of Lipinski’s rules.
Rule of 5 only addresses a narrow slice of the full gamut of hurdles a compound must pass before it can become a drug. In addition, it relies on “hard” thresholds: a compound with a molecular weight of 499 satisfies the MWt rule but a compound with a molecular weight of 501 violates it.
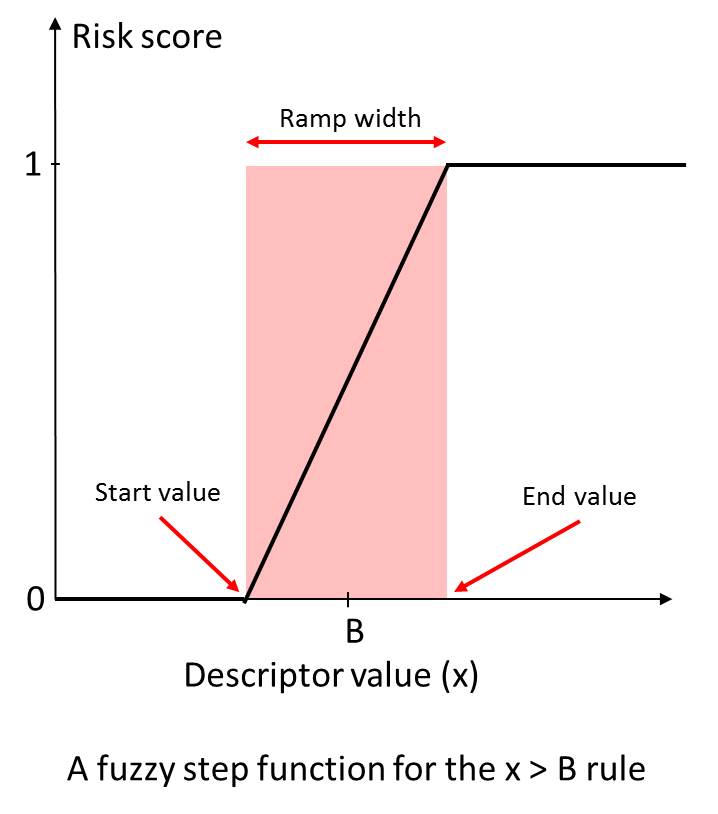
We calculated a broad range of relevant molecular descriptors and ADMET property predictions for the focused subset of WDI and identified “soft” threshold ranges for each along the lines suggested by (Petit; 2012) such that approximately 85% of the compounds in the data set satisfy them completely and somewhat less than 10% violate them completely. The former contribute nothing to the overall Risk, whereas the latter contribute the full amount (weight) specified for the corresponding rule. Predictions falling in the gray area in between contribute fractional amounts to the Risk Score. The concept is illustrated on the left.
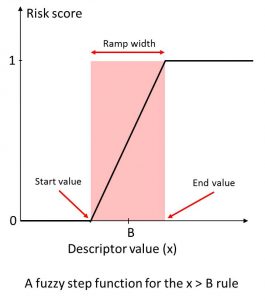 An illustration of “soft” thresholds for an inequality rule. The score starts increasing linearly from 0 at “start value” of the descriptor in the neighborhood of the boundary B, and reaches 1 at the “end value” value of the descriptor.
An illustration of “soft” thresholds for an inequality rule. The score starts increasing linearly from 0 at “start value” of the descriptor in the neighborhood of the boundary B, and reaches 1 at the “end value” value of the descriptor.
Highly correlated criteria were combined into single rules using Boolean operators. The rules for identifying overly large structures, for example, is:
size (Sz): MWt > [450,550] OR N_Atoms > [32,38] OR MolVol > [475,550] OR N_Bonds > [35,41]
where the values within the brackets indicate the boundaries of threshold regions. The Sz rule includes four individual criteria, all of which use the “>” relational operator. Predictions falling below the minimum threshold values contribute nothing to the Risk, whereas predictions above the maximum contribute 1 violation “point”. Intermediate values represent intermediate risks: a compound of molecular weight 500 violates the first criterion and so would represent an incremental Risk of 0.5 points for that criterion. Logical operators such as ORs and ANDs can also be included in the rules. The combined points from the criteria making up a rule then yield an overall value between 0 and 1, which is multiplied by the weight assigned to the rule as a whole.
The overall ADMET_Risk is the sum of three risks:
- Absn_Risk – risk of low fraction absorbed (PCB Module models)
- CYP_Risk – risk of high CYP metabolism (MET Module models)
- TOX_Risk – toxicity related risks (TOX Module models)
Two additional pharmacokinetic risks (high plasma protein binding and high steady-state volume of distribution) are also included in the ADMET_Risk score.
References
Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. “Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings.” Adv Drug Delivery Rev. 1997; 23:3-25.
Petit J, Meurice N, Kaiser C, Maggiora G. “Softening the Rule of Five – where to draw the line?” Bioorg Med Chem. 2012; 20:5343-5351.